

Special Offers
100% Performance Guaranteed
Key Specifications Table
| Species Reactivity | Key Applications | Host | Format | Antibody Type |
|---|---|---|---|---|
| B, H | IP, WB, IHC, IH(P) | M | Ascites | Monoclonal Antibody |
| Description | |
|---|---|
| Catalogue Number | MAB1680 |
| Replaces | CBL229 |
| Brand Family | Chemicon® |
| Trade Name |
|
| Description | Anti-Filamin A Antibody, clone TI10 |
| Alternate Names |
|
| Background Information | Filamin is a structural protein that forms flexible cross-links between two actin filaments. Filamin is a homodimer of polypeptide chains each joined to the other at one end with an actin binding site ath the other. It is present in smooth muscle, fibroblasts, platelets and lymphocytes. |
| Product Information | |
|---|---|
| Format | Ascites |
| Control |
|
| Presentation | Ascites. Liquid |
| Quality Level | MQ100 |
| Applications | |
|---|---|
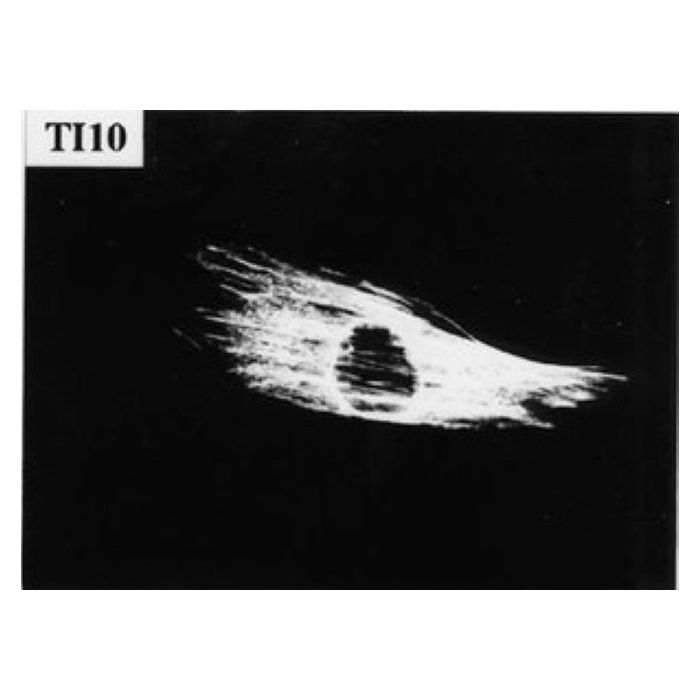
| Application | Detect Filamin A using this Anti-Filamin A Antibody, clone TI10 validated for use in IP, WB, IH, IH(P). |
| Key Applications |
|
| Application Notes | Immunoblotting: 1:250 to 1:1000 Immunoprecipitation:Suggested lysis buffer is PBS with 0.5% triton X-100 with proteinase inhibitors (note for full length filamin include calpain inhibitors). 5 microliters of antibody for every 300μL of cell lysate (3-5mg/ml total protein is suggested). Incubation is 4 hours RT or overnight 4C; Protein A/G agarose beads or rabbit anti-mouse secondary capture antibody is recommended for best recovery. 4-10% acrylamide gels are recommended for full length filamin or the 80kDa fragement visualization. Immunofluorescence: 1:50 to 1:200 using standard ABC technique. Suitable for staining of paraffin embedded sections (lower dilutions). High temperature citrate buffer antigen retrieval technique recommended. Optimal working dilutions must be determined by end user. |
| Biological Information | |
|---|---|
| Immunogen | Human platelet protein |
| Clone | TI10 |
| Host | Mouse |
| Specificity | Human filamin (actin-bidning protein). Recognizes unprocessed (270-280 kDa) and the C-terminal 90 kDa calpain cleavage fragment of filamin (Aakhus, 1992). |
| Isotype | IgG1 |
| Species Reactivity |
|
| Antibody Type | Monoclonal Antibody |
| Entrez Gene Number |
|
| Entrez Gene Summary | Actin-binding protein, or filamin, is a 280-kD protein that crosslinks actin filaments into orthogonal networks in cortical cytoplasm and participates in the anchoring of membrane proteins for the actin cytoskeleton. Remodeling of the cytoskeleton is central to the modulation of cell shape and migration. Filamin A, encoded by the FLNA gene, is a widely expressed protein that regulates reorganization of the actin cytoskeleton by interacting with integrins, transmembrane receptor complexes, and second messengers.[supplied by OMIM] |
| Gene Symbol |
|
| Non-Reactive Species |
|
| UniProt Number |
|
| UniProt Summary | FUNCTION: SwissProt: P21333 # Promotes orthogonal branching of actin filaments and links actin filaments to membrane glycoproteins. Anchors various transmembrane proteins to the actin cytoskeleton and serves as a scaffold for a wide range of cytoplasmic signaling proteins. Interaction with FLNA may allow neuroblast migration from the ventricular zone into the cortical plate. Tethers cell surface- localized furin, modulates its rate of internalization and directs its intracellular trafficking (By similarity). SIZE: 2647 amino acids; 280739 Da SUBUNIT: Interacts with PDLIM2 (By similarity). Homodimer. Interacts with FLNB, FURIN, HSPB7, INPPL1, KCND2, MYOT, MYOZ1, ARHGAP24, PSEN1 and PSEN2. Interacts also with various other binding partners in addition to filamentous actin. SUBCELLULAR LOCATION: Cytoplasm, cell cortex. TISSUE SPECIFICITY: Ubiquitous. DOMAIN: SwissProt: P21333 Comprised of a NH2-terminal actin-binding domain, 24 internally homologous repeats and two hinge regions. Repeat 24 and the second hinge domain are important for dimer formation. PTM: Phosphorylated upon DNA damage, probably by ATM or ATR (By similarity). Phosphorylation extent changes in response to cell activation. & The N-terminus is blocked. DISEASE: SwissProt: P21333 # Defects in FLNA are the cause of periventricular nodular heterotopia 1 (PVNH1) [MIM:300049]; also called nodular heterotopia, bilateral periventricular (NHBP or BPNH). PVNH1 is an X-linked developmental dominant disorder in which many neurons fail to migrate into the cerebral cortex. They remain as nodules lining the ventricular surface. In heterozygous females these neurons presumably represent those cells that, after X-chromosome inactivation, contain the active X chromosome with the filamin mutation. Most hemizygous affected males die early during embryogenesis, whereas heterozygous females have normal intelligence but suffer from seizures and various manifestations outside the central nervous system, especially related to the vascular system. This implies that essential embryonic cell migration can only occur in FLNA-expressing cells. & Defects in FLNA are the cause of periventricular nodular heterotopia 4 (PVNH4) [MIM:300537]; also known as periventricular heterotopia Ehlers-Danlos variant. PVNH4 is characterized by nodular brain heterotopia, joint hypermobility and development of aortic dilatation in early adulthood. & Defects in FLNA are the cause of otopalatodigital syndrome type 1 (OPD1) [MIM:311300]. OPD1 is an X-linked dominant multiple congenital anomalies disease mainly characterized by a generalized skeletal dysplasia, mild mental retardation, hearing loss, cleft palate, and typical facial anomalies. OPD1 belongs to a group of X-linked skeletal dysplasias known as oto-palato- digital syndrome spectrum disorders that also include OPD2, Melnick-Needles syndrome (MNS), and frontometaphyseal dysplasia (FMD). Remodeling of the cytoskeleton is central to the modulation of cell shape and migration. FLNA is a widely expressed protein that regulates re-organization of the actin cytoskeleton by interacting with integrins, transmembrane receptor complexes and second messengers. Males with OPD1 have cleft palate, malformations of the ossicles causing deafness and milder bone and limb defects than those associated with OPD2. Obligate female carriers of mutations causing both OPD1 and OPD2 have variable (often milder) expression of a similar phenotypic spectrum. & Defects in FLNA are the cause of otopalatodigital syndrome type 2 (OPD2) [MIM:304120]; also known as cranioorodigital syndrome. OPD2 is a congenital bone disorder that is characterized by abnormally modeled, bowed bones, small or absent first digits and, more variably, cleft palate, posterior fossa brain anomalies, omphalocele and cardiac defects. & Defects in FLNA are the cause of frontometaphyseal dysplasia (FMD) [MIM:305620]. FMD is a congenital bone disease characterized by supraorbital hyperostosis, deafness and digital anomalies. & Defects in FLNA are the cause of Melnick-Needles syndrome (MNS) [MIM:309350]. MNS is a severe congenital bone disorder characterized by typical facies (exophthalmos, full cheeks, micrognathia and malalignment of teeth), flaring of the metaphyses of long bones, s-like curvature of bones of legs, irregular constrictions in the ribs, and sclerosis of base of skull. & Defects in FLNA are associated with cerebrofrontofacial syndrome [MIM:608578]. This syndrome consists of a phenotype of male PVNH, with relatively normal development, no epilepsy or other neurological abnormality, severe constipation, and facial dysmorphism and without a discernible skeletal phenotype. & Defects in FLNA are a cause of X-linked congenital idiopathic intestinal pseudoobstruction (CIIPX) [MIM:300048]. CIIPX is caused by severe abnormality of gastrointestinal motility either due to primary qualitative defects of enteric ganglia and nerve fibers, or secondary to a variety of conditions, such as myopathies, inflammatory or autoimmune diseases, drug toxicity, ischemia, or viral infections. CIIPX is diagnosed by radiological, surgical, or manometric evidence of abnormal bowel motility causing intestinal obstruction in the absence of any mechanical occlusion. SIMILARITY: Belongs to the filamin family. & Contains 1 actin-binding domain. & Contains 2 CH (calponin-homology) domains. & Contains 24 filamin repeats. |
| Product Usage Statements | |
|---|---|
| Usage Statement |
|
| Storage and Shipping Information | |
|---|---|
| Storage Conditions | Maintain frozen at -20°C for up to 12 months in undiluted aliquots. Avoid repeated freeze/thaw cycles. |
| Packaging Information | |
|---|---|
| Material Size | 100 µL |